Atomistic modeling techniques are a class of models that, unlike continuum models, incorporate atomic-level information to simulate macroscopic material properties. Atomistic modeling considers atoms as its smallest entity and the most basic building blocks of materials. The most fundamental atomistic models, based on so-called ‘first principles,’ take into account both atomic nuclei and electron density’s spacial variation to predict materials properties, a priori. Our group utilizes plane-wave basis-set density functional theory (PW-DFT) as a first principles model to investigation fundamental properties of materials using the VASP. Molecular dynamics (MD), often thought of as a force field model, in essence, models the forces felt/exerted on/by each atom to determine macroscopic material properties and atomic data. These methods, along with many other models, are useful tools to elucidate fundamental mechanisms which give rise to intrinsic material properties. These intrinsic material properties can be correlated with experimental high resolution TEM spectra to validate mechanisms which give rise to specific structure-function relationships.
Density Functional Theory
Density functional theory is just one of the tools our group utilizes to interpret atomic resolution images and spectra obtained using the TEM facilities at ASU. Currently, our group uses the Vienna Ab-initio Software Package (VASP) to study the relationship between grain boundary character and ionic conductivity in solid oxide fuel cell electrolytes (SOFC). This work is a continuation of the work done by several group members, both experimental and theoretical. The experimental research was performed by a previous graduate student, William Bowman, who worked on correlating nanoscale grain boundary composition with electrical conductivity in ceria. The theoretical work was done by a post-doctoral research assistant, Pratik Dholabhai, who worked with kinetic lattice monte carlo (KLMC) and DFT to optimize ionic conductivity in ceria through doping. Our groups current theorist is a graduate research assistant, Tara Boland, who is working to correlate grain boundary character with ionic conductivity in ceria grain boundaries.
Molecular Dynamics
LAMMPS is our groups molecular dynamics (MD) simulation package, provided by Sandia National Laboratory (open source). MD provides a scale-able method to investigate more realistic length and time scales for material systems. Grain boundaries in particular pose computational challenges due to the many atom-system. MD easily simulates many grain boundary orientations allowing the construction of the potential energy surface for grain boundary structures.
DFT is used for fine structural refinements and electronic information due to the large computational resources needed to simulate these materials. Various other methods such as image simulation, and spectral simulation using FEFF, are implemented to correlate the structure with certain functionalities of materials.
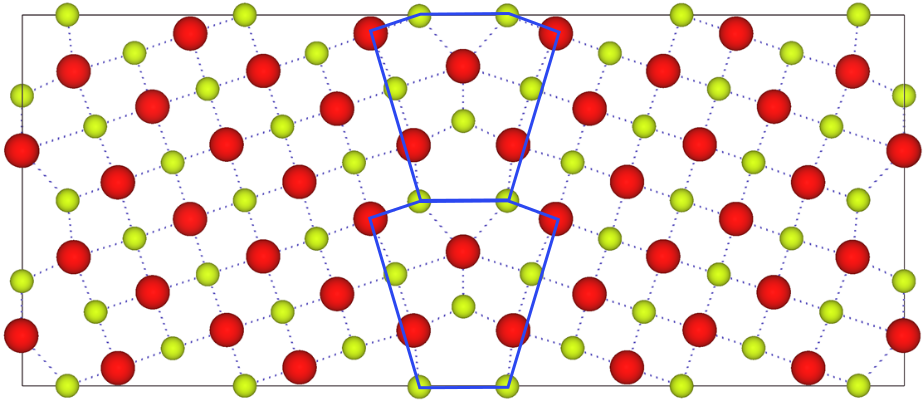
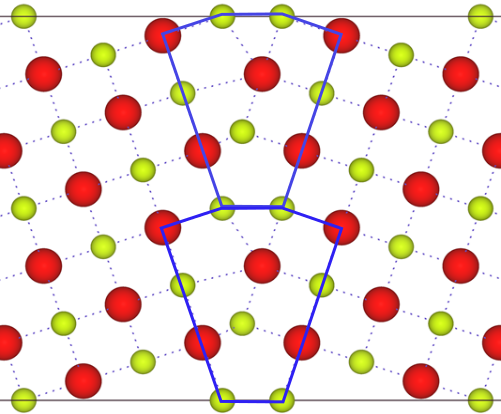 The figure to the left is the Σ5(210)/[001] symmetric tilt grain boundary (GB) before relaxing the structure. Each supercell has a volume of 28.80 x 12.09986 x 5.4112 Å3. The supercell has 2 identical GB’s per supercell. Upon relaxation, the GB core increases in volume. A total of 0.6 Å in the x direction was added to the cell per GB.
The figure to the left is the Σ5(210)/[001] symmetric tilt grain boundary (GB) before relaxing the structure. Each supercell has a volume of 28.80 x 12.09986 x 5.4112 Å3. The supercell has 2 identical GB’s per supercell. Upon relaxation, the GB core increases in volume. A total of 0.6 Å in the x direction was added to the cell per GB.
DFT is capable of more accurate structural refinement which is essential for the accuracy of image and spectral simulations.